.
Natural Peptides |
Beginner
It is a simple interface in which user needs to give an input sequence and his/her email address. User can also select different advance options, which include Simulation Time, Peptide Environment, Download topology files, Cluster Analysis, Download whole trajectory, Energy and RMS graphs. These advanced options assist the user in further analysis of their peptides (Figure 1).
|
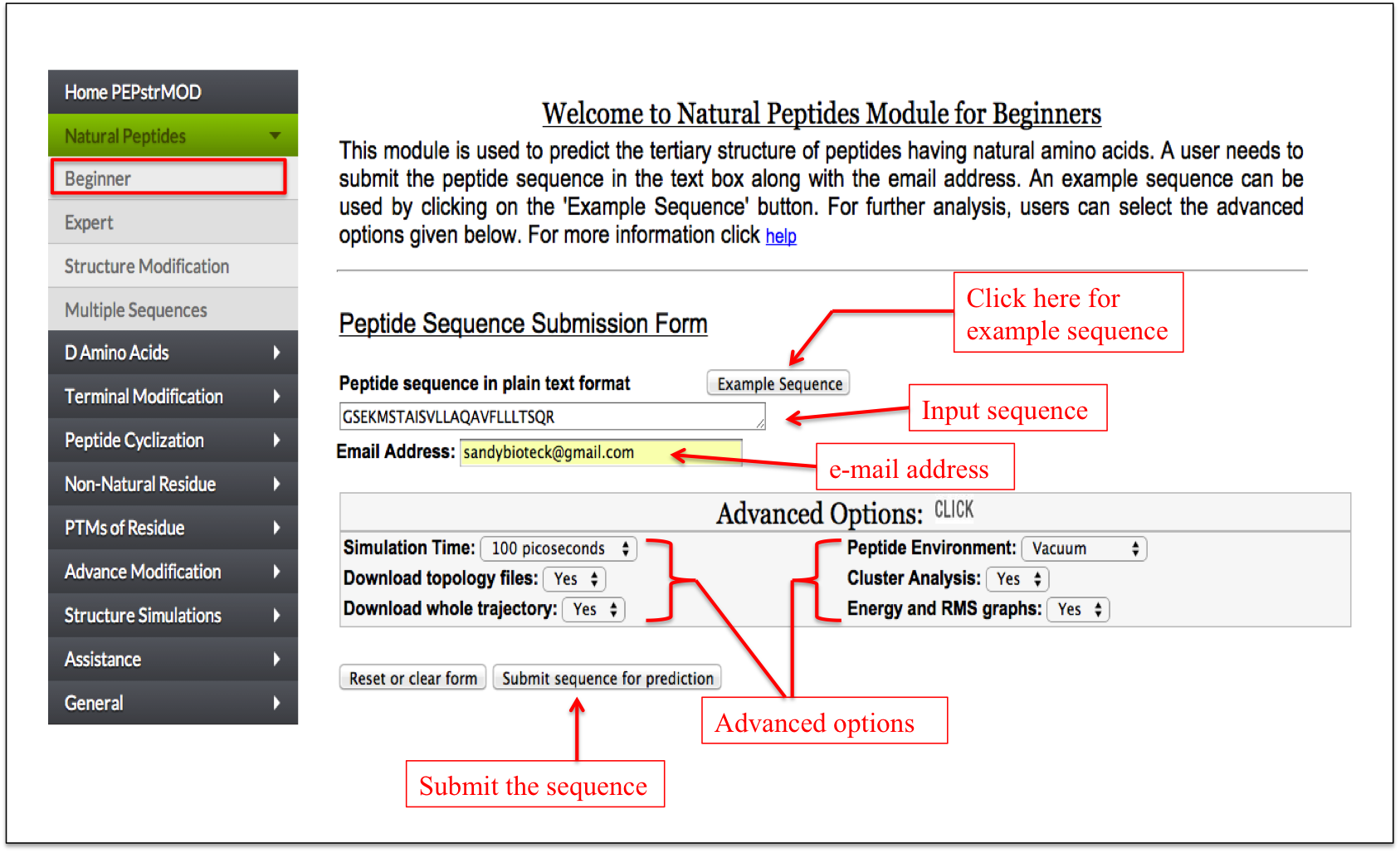
Figure 1. Graphical representation showing how to use beginner sub module of natural peptide module of PEPstrMOD. |
|
Expert
This facility allows a user to design his/her peptide using defined secondary structure (helix/sheet) or backbone torsion angles phi and psi. A user can select the region of a peptide to adopt a particular secondary structure or any shape by input of phi and psi torsion angles. The interface is designed in a very user-friendly manner, which makes very easy to design a peptide (Figure 2A-B).
|

Figure 2A. Snapshot of expert sub module of natural peptide module of PEPstrMOD and labels to assist users to use the service effectively. At this step a user only needs to input the sequence and click on the link to go to the next step. |
|
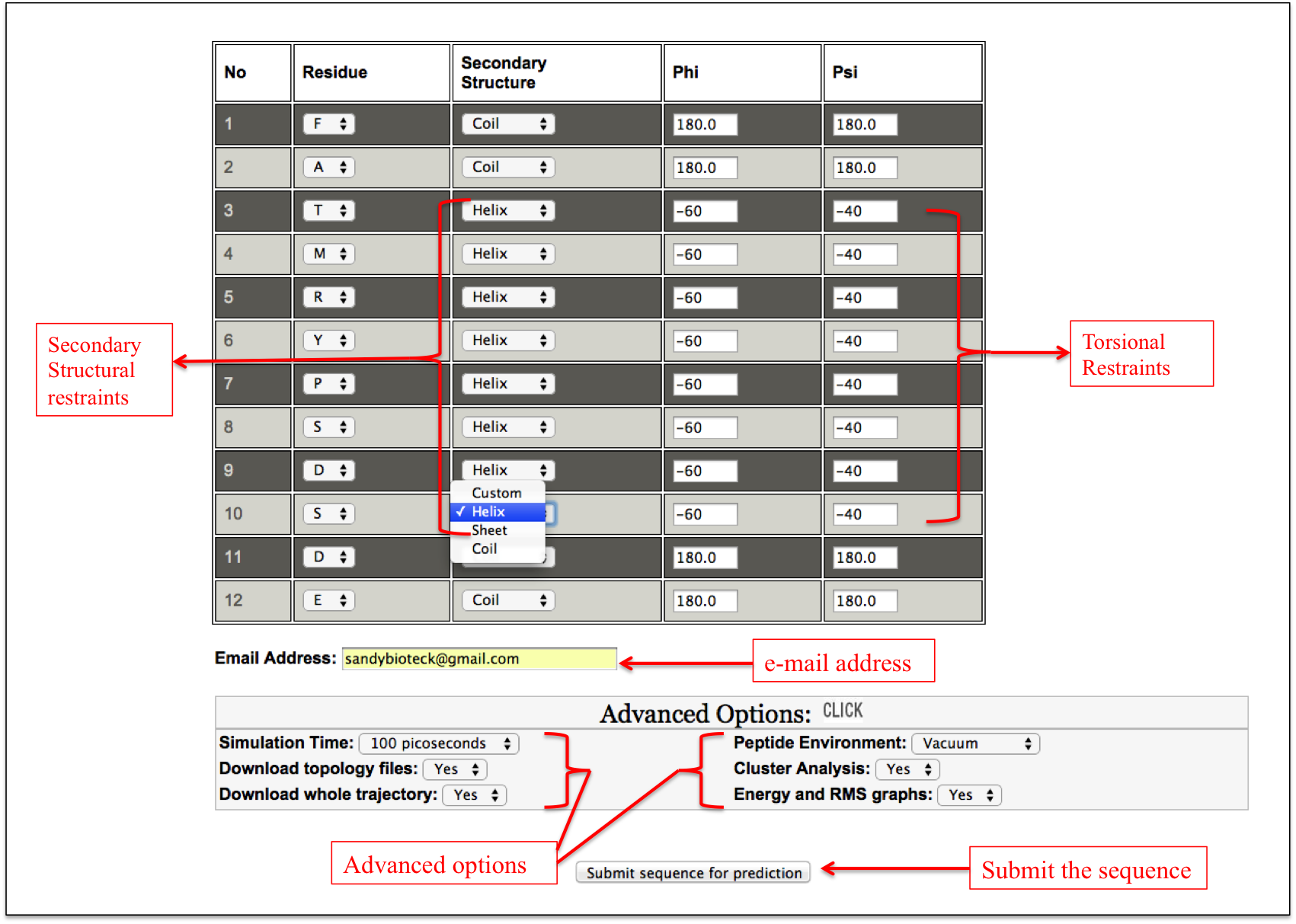
Figure 2B. This snapshot is the extension of the Expert sub module of “Natural Peptide” module of PEPstrMOD. The peptide sequence submitted in the previous step is displayed here in the tabular format and a user can apply secondary structural or torsion angle restraints to model his/her peptide. In this example peptide shown in this figure, secondary structural (helical) restraints are applied to the region from 3rd to 10th residue of the peptide. |
|
Structure Modification
If a user already has a peptide tertiary structure in PDB format and want to further design the region of the peptide with his/her intuition of backbone torsion angles or specific secondary structure or a mutation at a specific position, then this option allows him/her to do this task in a very easy manner. This option will be very helpful in user aided designing of the peptide tertiary structure (Figure 3).
|
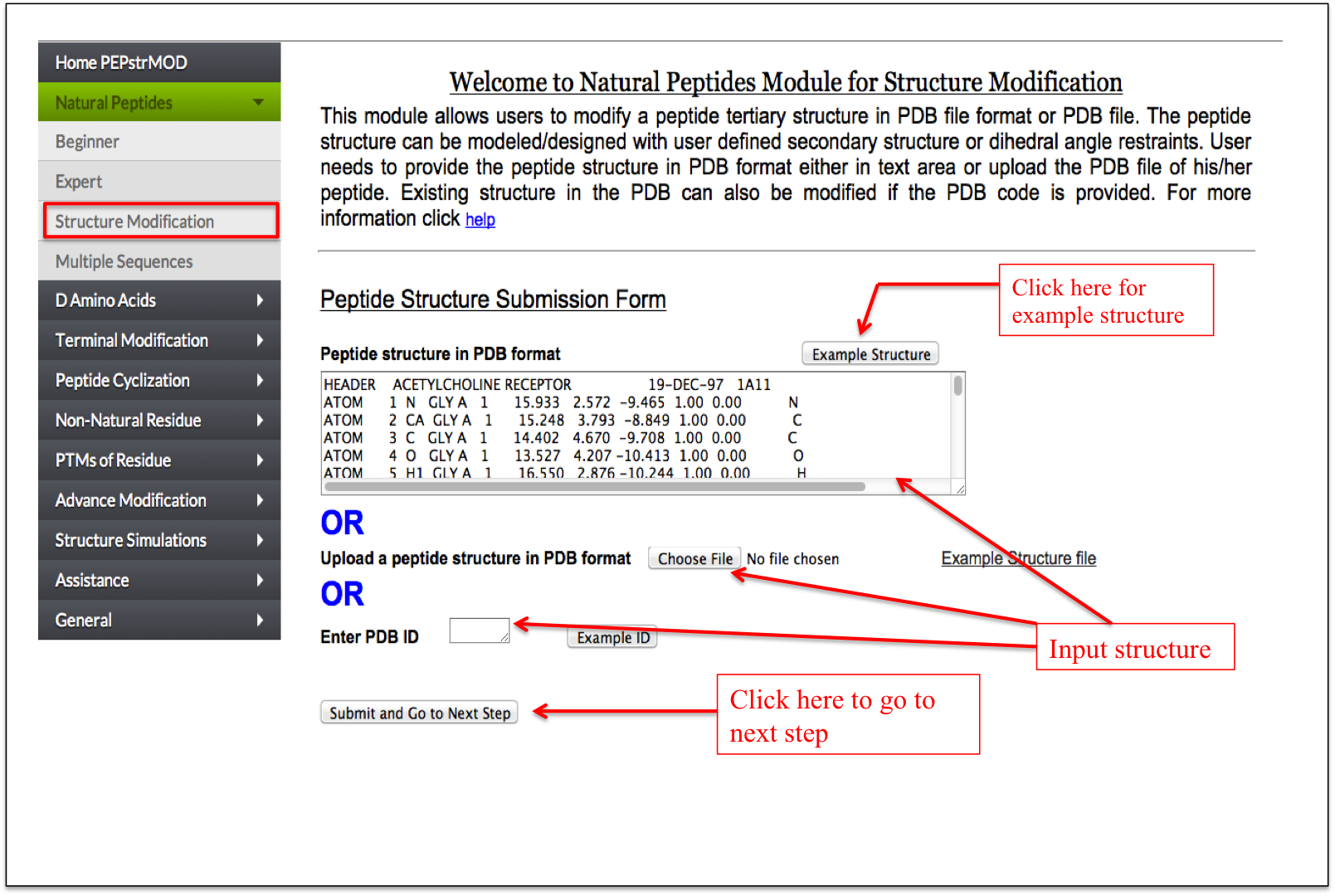
Figure 3. Snapshot of the “Structure Modification” sub module of the “Natural Peptide” module of the PEPstrMOD showing how to use the features and options effectively. |
|
Multiple Sequences
If a user has multiple peptides, he/she can process such peptides with this sub module. A user needs to give the multiple peptide sequences in a single line format (i.e. each peptide sequence in one line) and his/her email address (Figure 4). Due to the limited computational power and to avoid heavy load on our server, we do not perform the molecular dynamics simulation of these peptides, which is the last step of PEPstrMOD. Instead we only perform energy minimization step on the initial predicted structure. The results (PDB structure file) for all the peptides is compressed and provided in a .tgz format. This can be extracted using following command in linux “tar –xzf file.tgz” or using the WinRAR utility in Window based operating system. The result is also provided in the tabular format in which the user can download individual peptide structure (Figure 4).
|
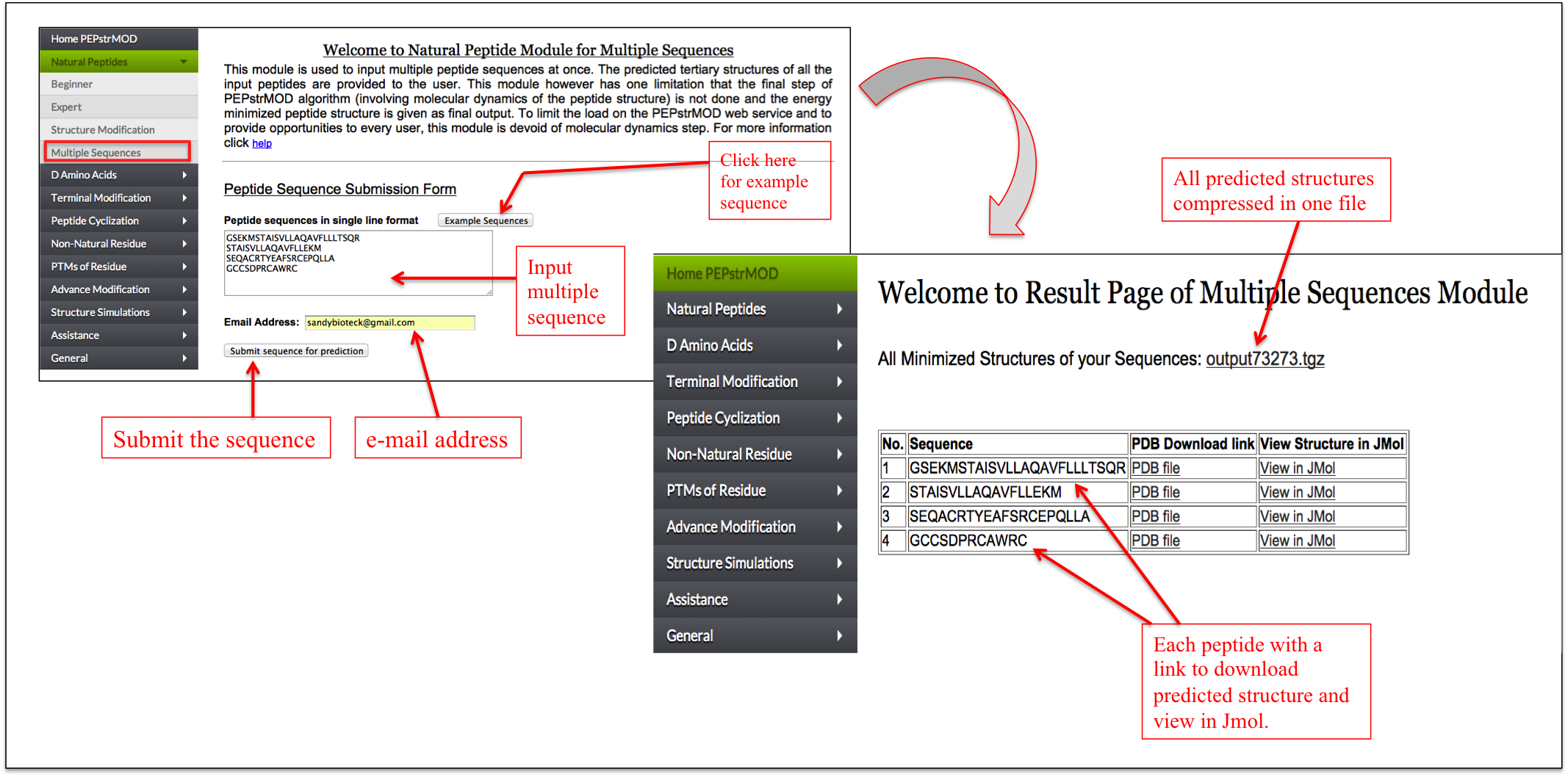
Figure 4. Snapshot of the Multiple Sequences sub module of the Natural Peptides module of PEPstrMOD. |
|
D Amino Acids |
Beginner
A user needs to input the peptide sequence and after submitting the sequence, a user friendly tabular interface is displayed with amino acid and its corresponding stereo-chemistry in drop down menu. A user can select either the Laevo (L) or the Dextro (D) stereo-chemistryof any amino acid in the peptide sequence and proceed further for the prediction of its tertiary structure.
|
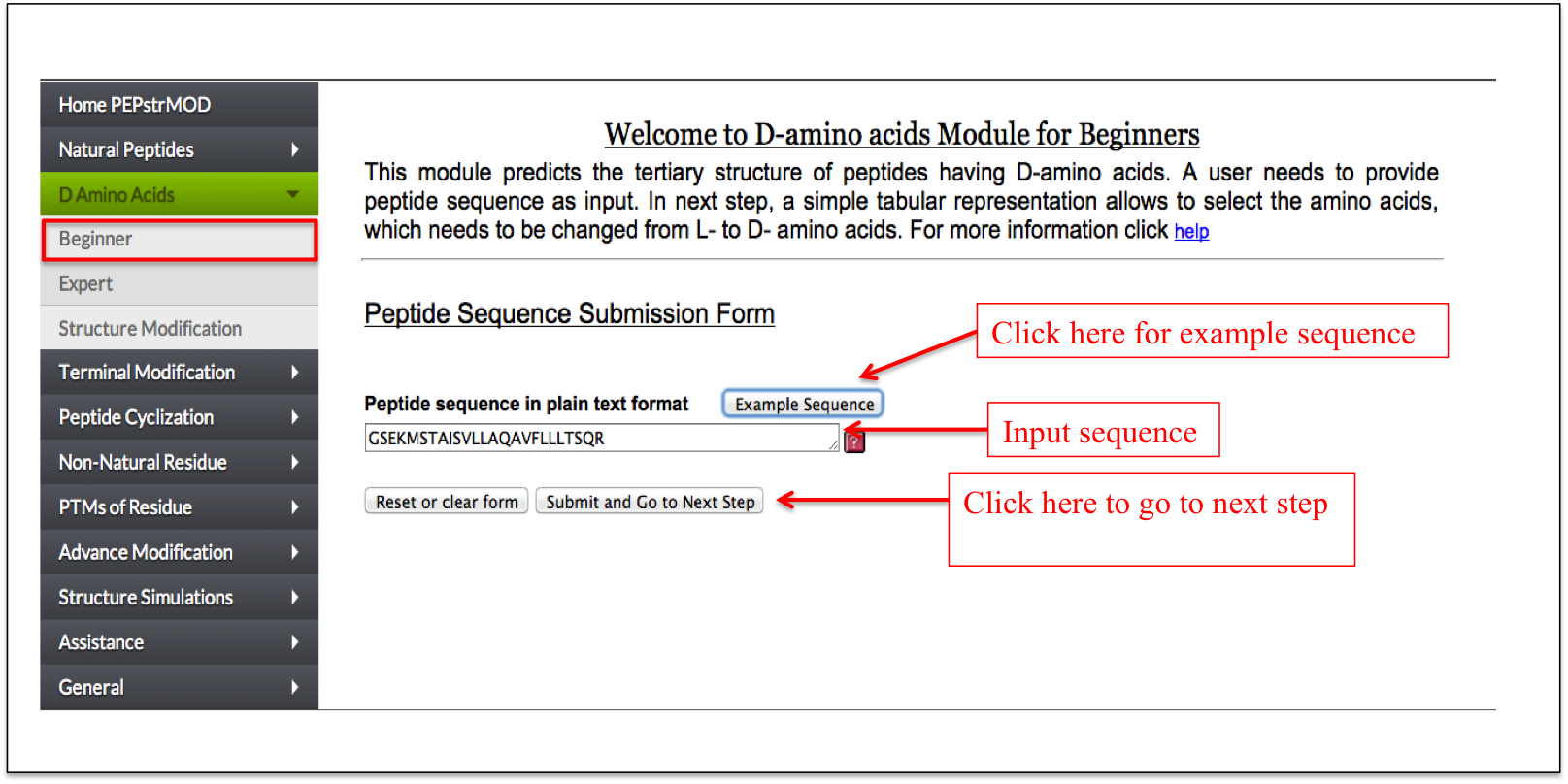
Figure 5A. Snapshot of Beginner sub module of “D-amino acid” module of PEPstrMOD labeling the inputs and options required for the peptide submission. After submission, the next step involves selection of residues and its L/D stereo-chemistry. |
|
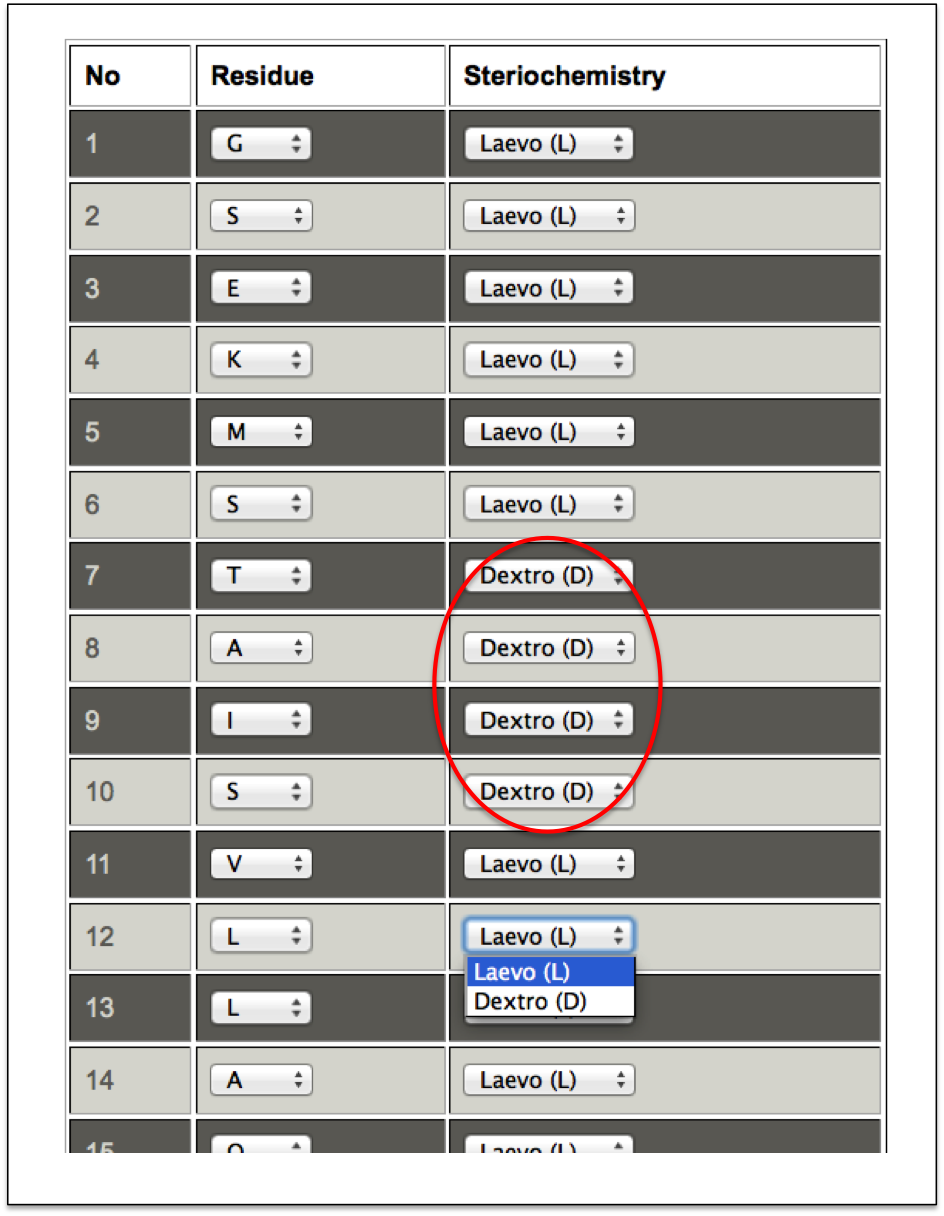
Figure 5B. This snapshot is the extension of the Beginner sub module of “D-amino acid” module of PEPstrMOD. Using the dropdown menu in front of each amino acid of the peptide sequence (submitted in the previous step), a user can select the amino acid to be either Leavo or Dextro. Residues selected with Dextro stereo-chemistry are labeled with red circle. |
|
Expert
This is a simple interface which allows a user to enter a peptide sequence with L-amino acids in capital letter and D-amino acids in small letter representation. Alternatively, a user can enter the peptide sequence with all amino acids in capital letter and specify the positions of amino acids in a text box which needs to be converted in D-amino acids.
|
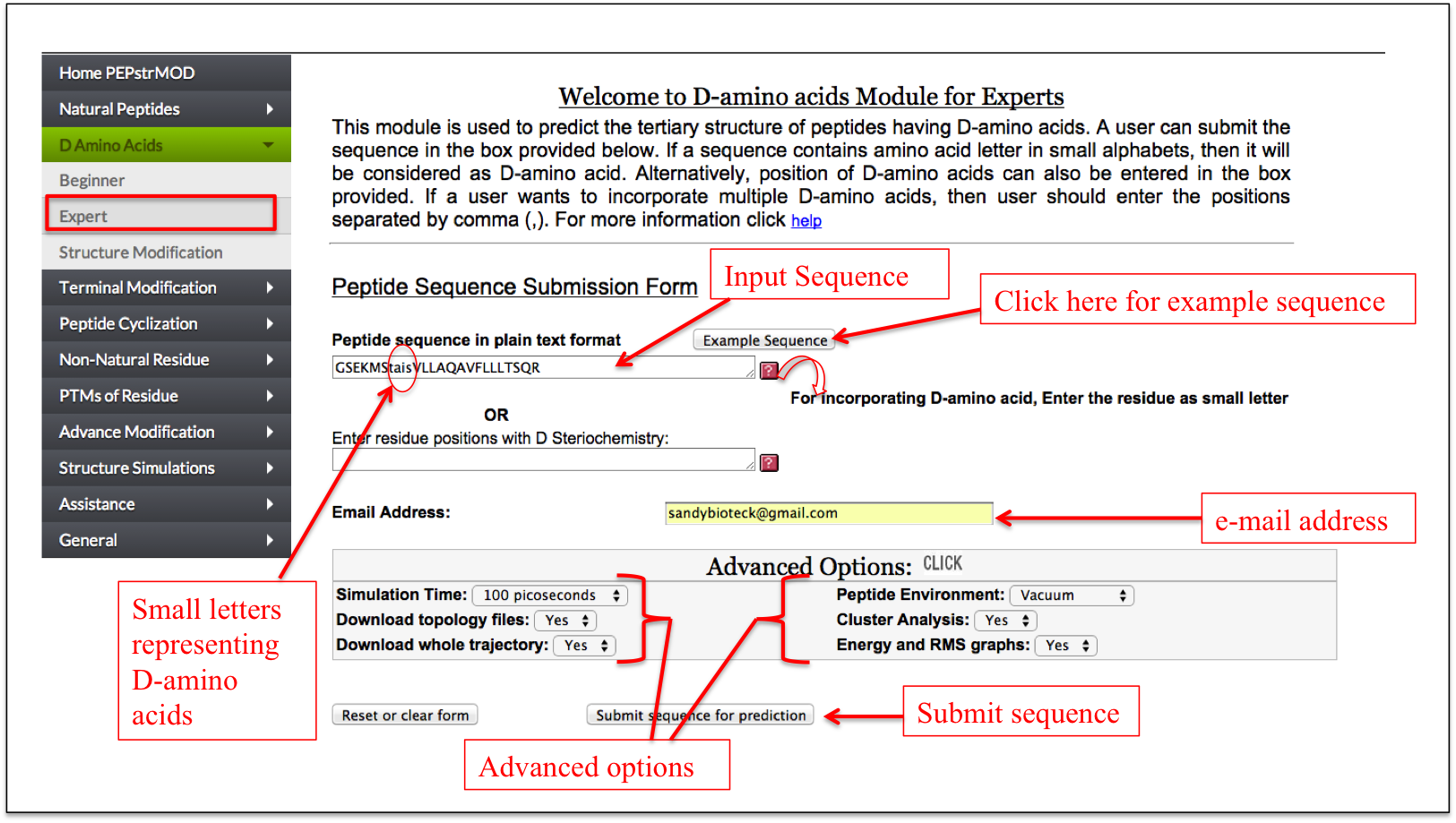
Figure 6A. Graphical representation of “Expert” sub module of “D Amino acid” module of PEPstrMOD. Residues “tais” are written in small letter representing D-amino acids and therefore are treated as such in prediction of the tertiary structure of the peptide. |
|

Figure 6B. This figure also represents “Expert” sub module of “D Amino acid” module of PEPstrMOD but rather than representing D-amino acids as small letter, their respective positions in the peptide sequence are given as input in the box provided. |
|
Structure Modification
This facility is same as described above with the additional option of incorporating D-amino acids at any position in the tertiary structure of the input peptide.
|
Terminal Modification |
Sequence Modification
If a user wants to block the N and C terminus of the peptide by capping these terminals with some chemical groups, he/she can perform the capping using this module of PEPstrMOD. A user can add acetylation at the N-terminus and amidation or n-methylamide groups at the C-terminus residue of the peptide respectively (Figure 7).
|
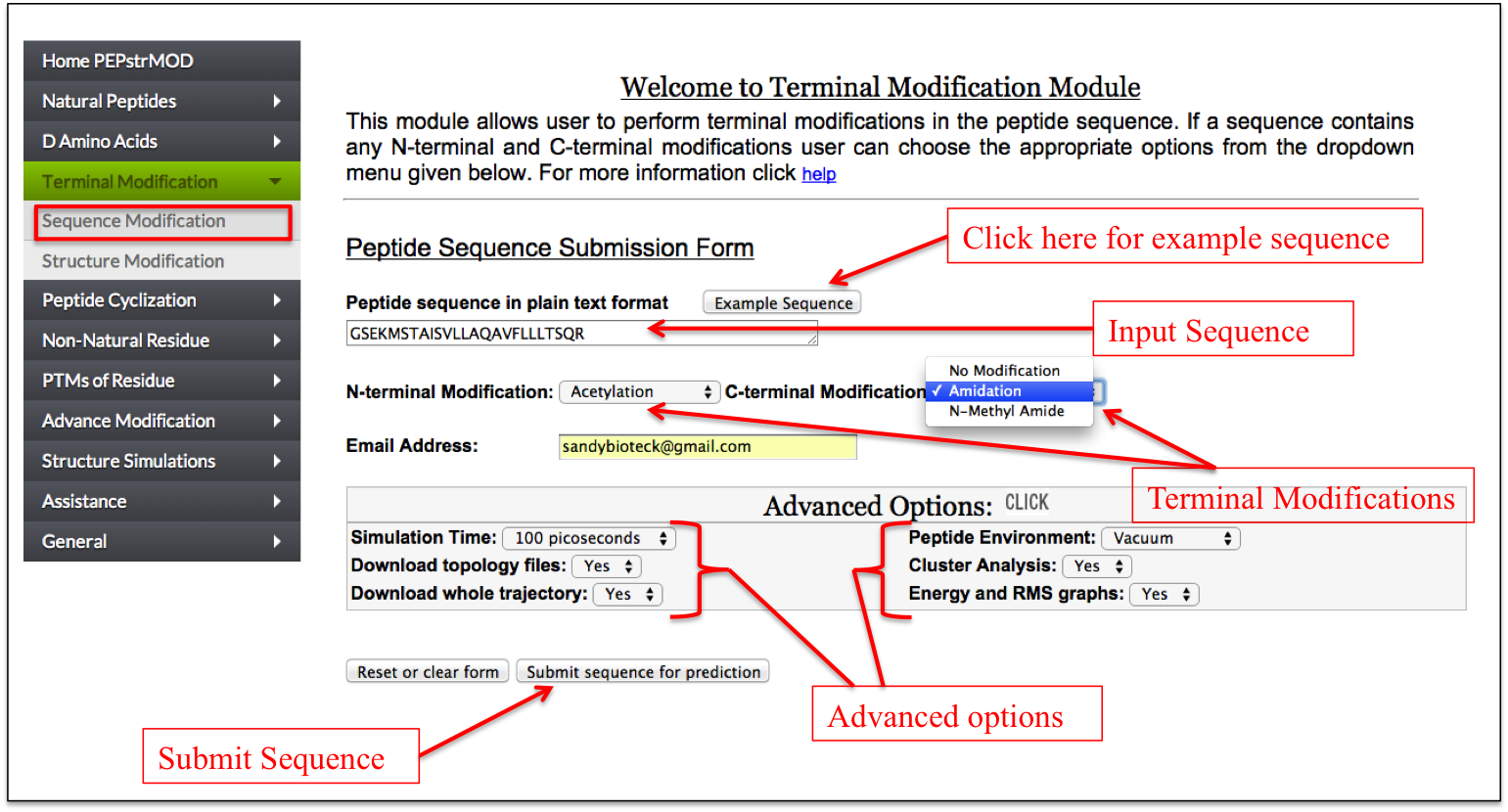
Figure 7. Graphical representation of the “Sequence Modification” sub module of “Terminal Modification” module of PEPstrMOD and labels showing how to input the peptide sequence along with selecting other options. |
|
Structure Modification
This facility is same as described in the Natural Peptides module of PEPstrMOD with additional option of incorporating N and C terminal modifications in the tertiary structure of the input peptide (Figure 8).
|
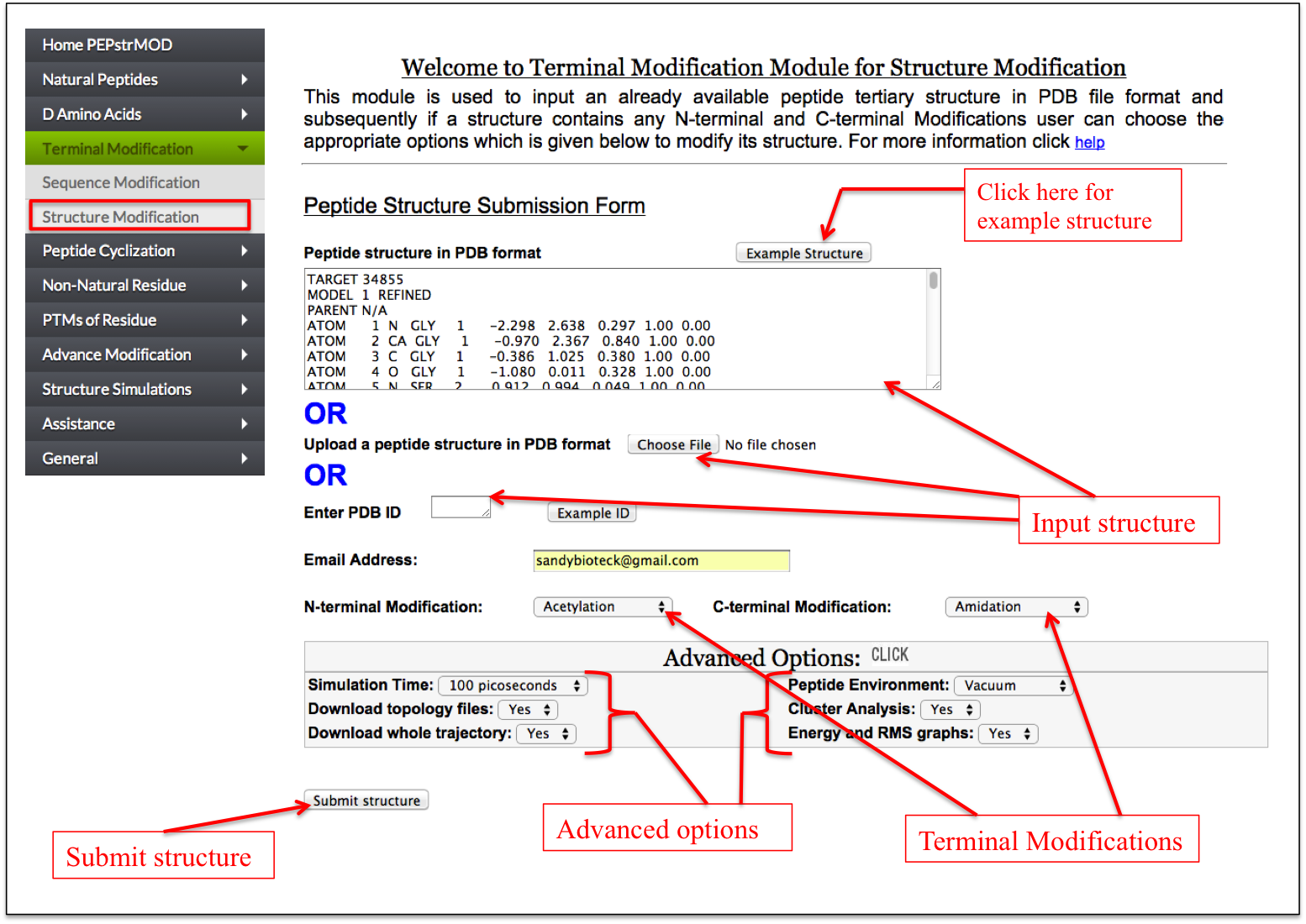
Figure 8. The snapshot of “Structure Modification” sub module of “Terminal Modification” module of PEPstrMOD showing the labels on how to use the module effectively. |
|
Peptide Cyclization |
N-C Cyclization
This page is designed to predict peptide structure with N-to-C terminal cyclization. The peptide is made cyclic by incorporating a peptide bond between Nitrogen atom of N-terminal residue and Carbon atom of C-terminal residue (Figure 9).
|
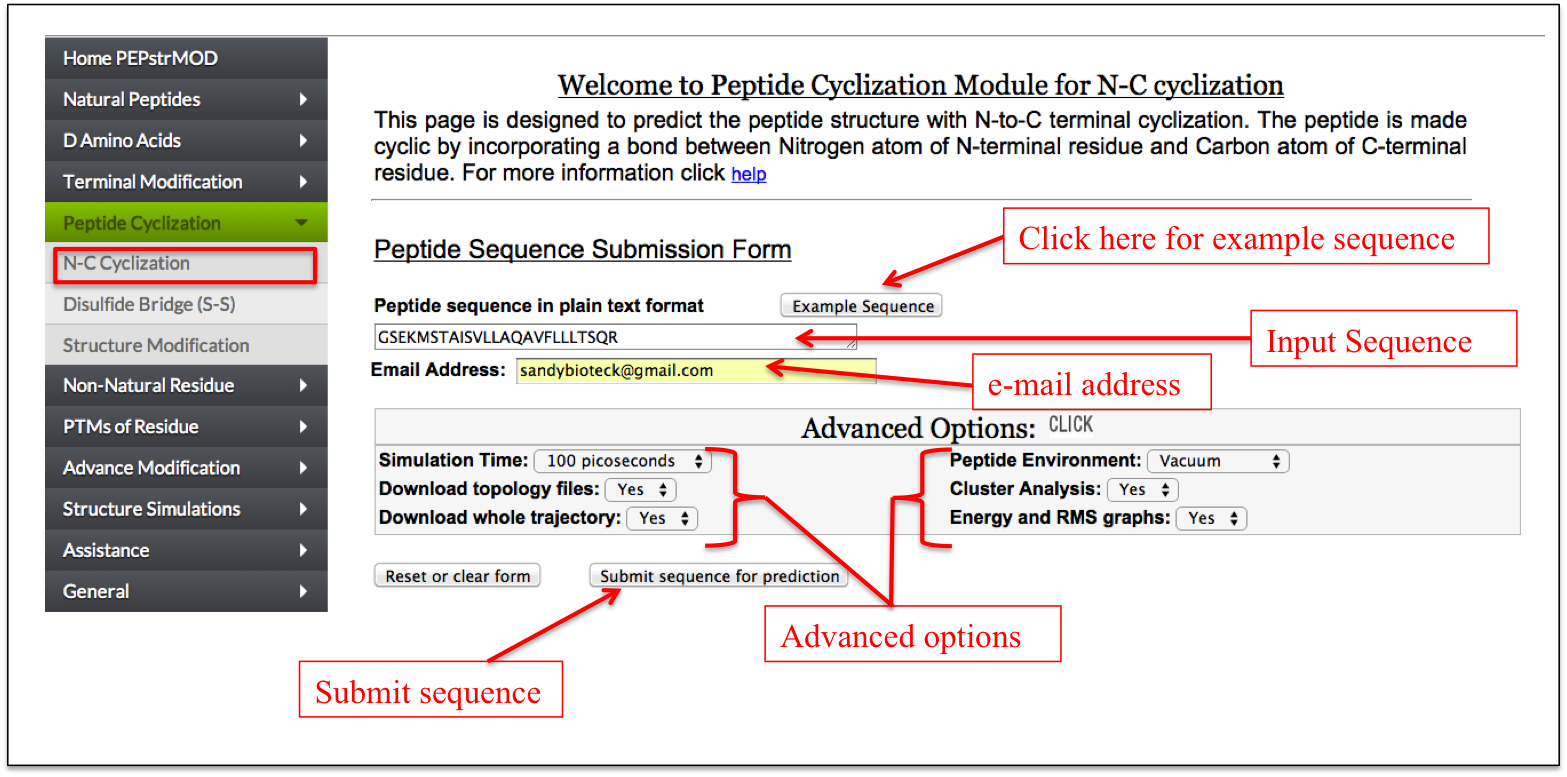
Figure 9. Labels showing the use of “N-C Cyclization” sub module of “Peptide Cyclization” module of PEPstrMOD. The structure predicted by this feature is shown in the next figure. |
|
Disulfide Bridge(S-S bonds)
This page is Designed so that user can add the Di-sulfide Bridge between two Cysteine residues. For example if you want to add S-S bond between cysteine at postion no. 5 and 10 then input numeric value one in the box provided in the front of both cysteine. If there is more than one S-S bond then input numeric value with respective increment (Figure 10).
|
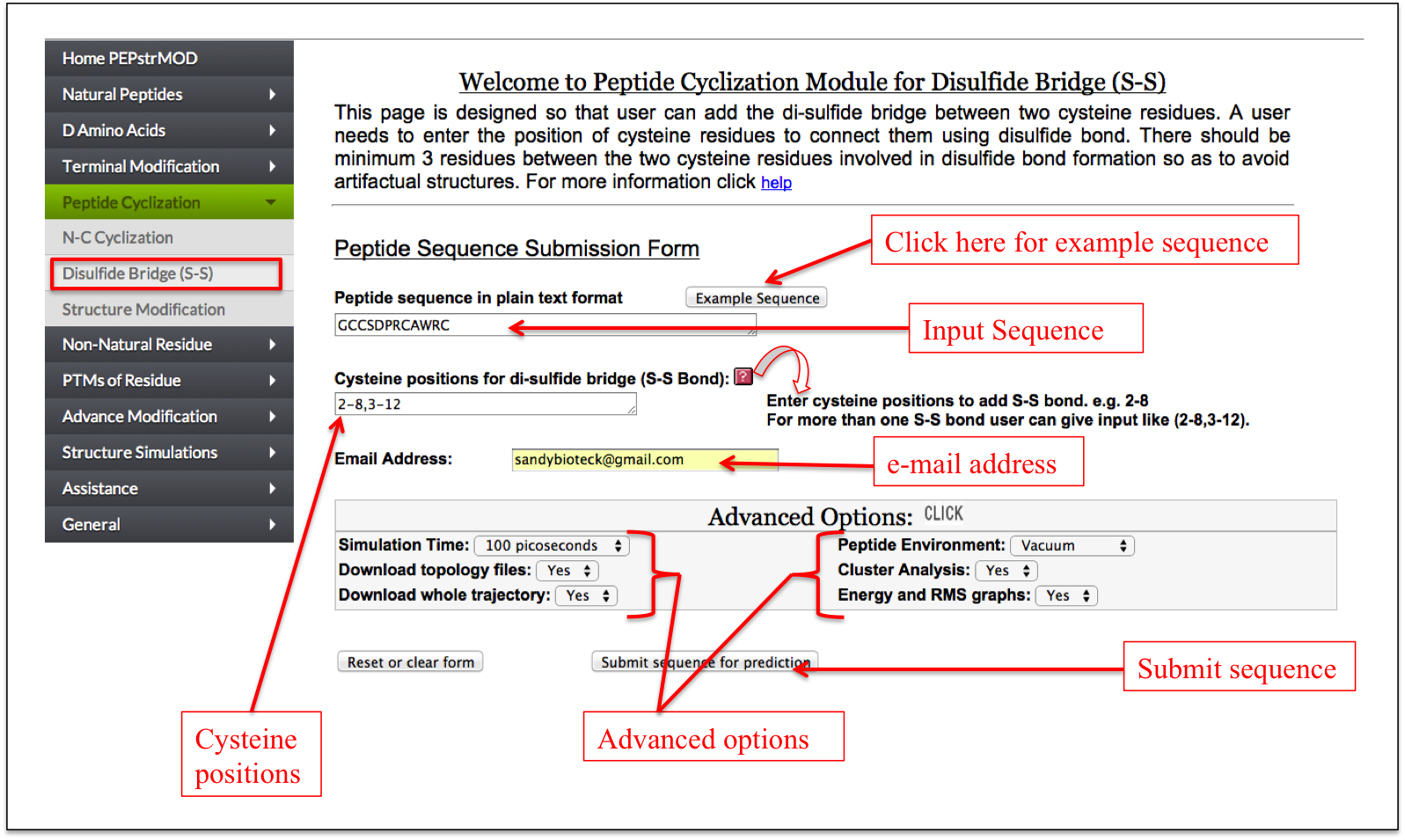
Figure 10. Graphical representation of the “Disulfide Bridge” sub module of the “Peptide Cyclization” module of the PEPstrMOD. The different labels show how to input the peptide sequence along with the cysteine positions, which needs to be joined by a disulfide bridge. |
|
Structure Modification
This facility is same as described in the Natural Peptides module of PEPstrMOD with additional option of incorporating disulfide bridges between cysteine residues in the tertiary structure of the input peptide.
|
Non-Natural Residue |
Beginner
This sub module provides an easy alternative to incorporate non-natural modifications in the peptide. At first step, a user only needs to enter the peptide sequence, and select either Forcefield_NCAA or SwissSideChain library of non-natural amino acids. After submission, a user friendly interface in the tabular form is displayed in which each amino acid of the input peptide sequence is present in one column and a drop down menu of all possible non-natural amino acids occur in the next column. A user can easily replace any amino acid of the peptide with any non-natural modification from the drop down menu (Figure 11A-B).
|

Figure 11A. Snapshot of “Beginner” sub module of the “Non-Natural Residue” module of PEPstrMOD with labels showing how to use its options effectively. The option to incorporate non-natural modification is provided in the next page as shown in the next figure. |
|
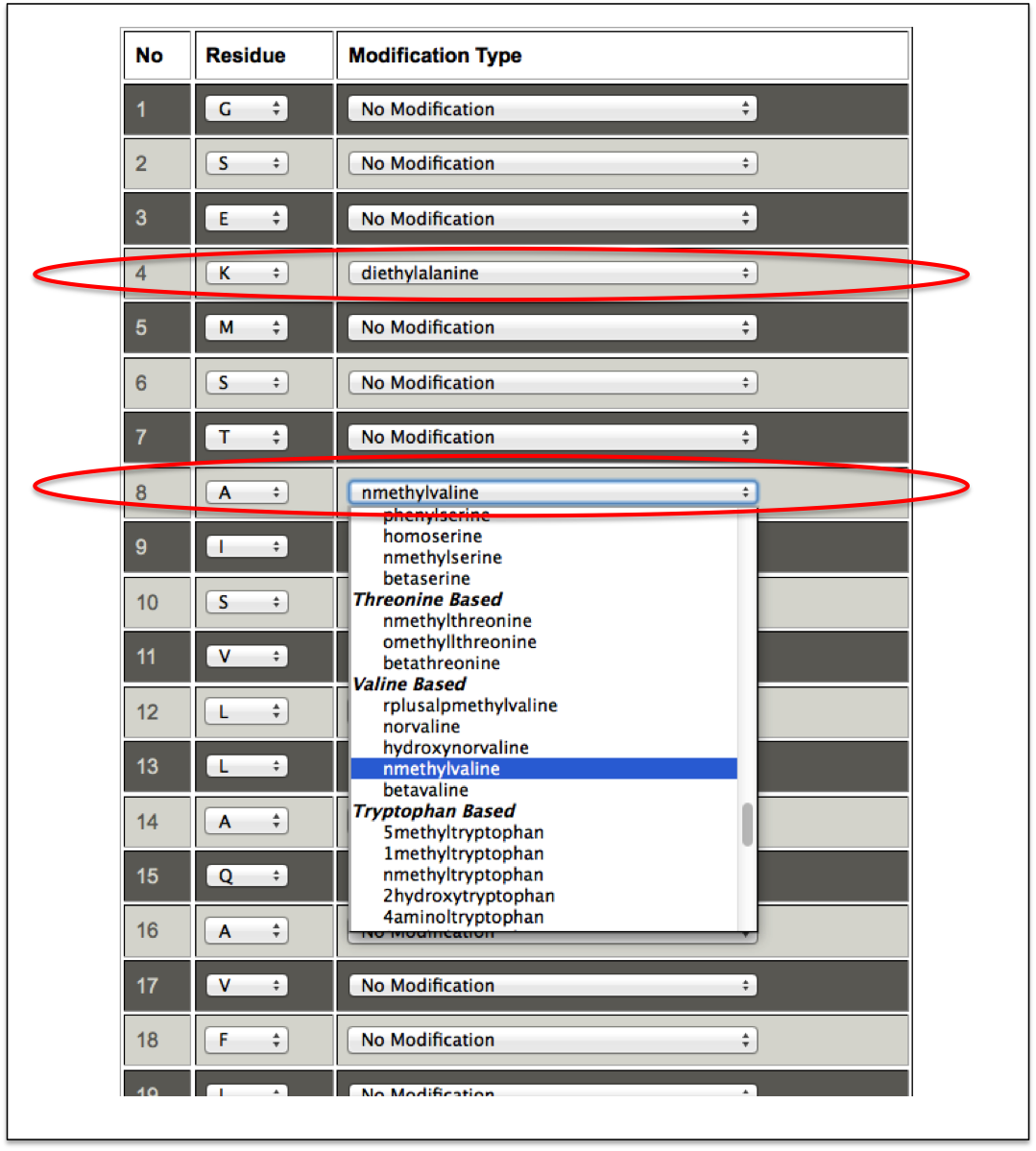
Figure 11B. This snapshot is the extension of the previous figure 11A in which peptide sequence was taken as input. In this step, non-natural modifications can be incorporated by using dropdown menu in front of amino acids of the input peptide sequence. In this figure the incorporated non-natural residues are labeled with red ellipse. |
|
Expert
It allows users to incorporate any non-natural amino acid at any position in the peptide in a single step. A user needs to input the sequence of his/her peptide and needs to select either Forcefield_NCAA or SwissSideChain library. After selecting a particular force field library, the respective non-natural amino acids are displayed in a drop down menu along with the natural amino acids from which their parameters are derived. For example, diethylalanine non-natural amino acid in the ForceField_NCAA library is similar to alanine and therefore all the non-natural amino acids (like diethylalanine, N-methylalanine etc.) similar to alanine are displayed in a drop down in front of alanine. In this way, a user can incorporate multiple non-natural amino acids at different positions in the peptide (Figure 12).
|
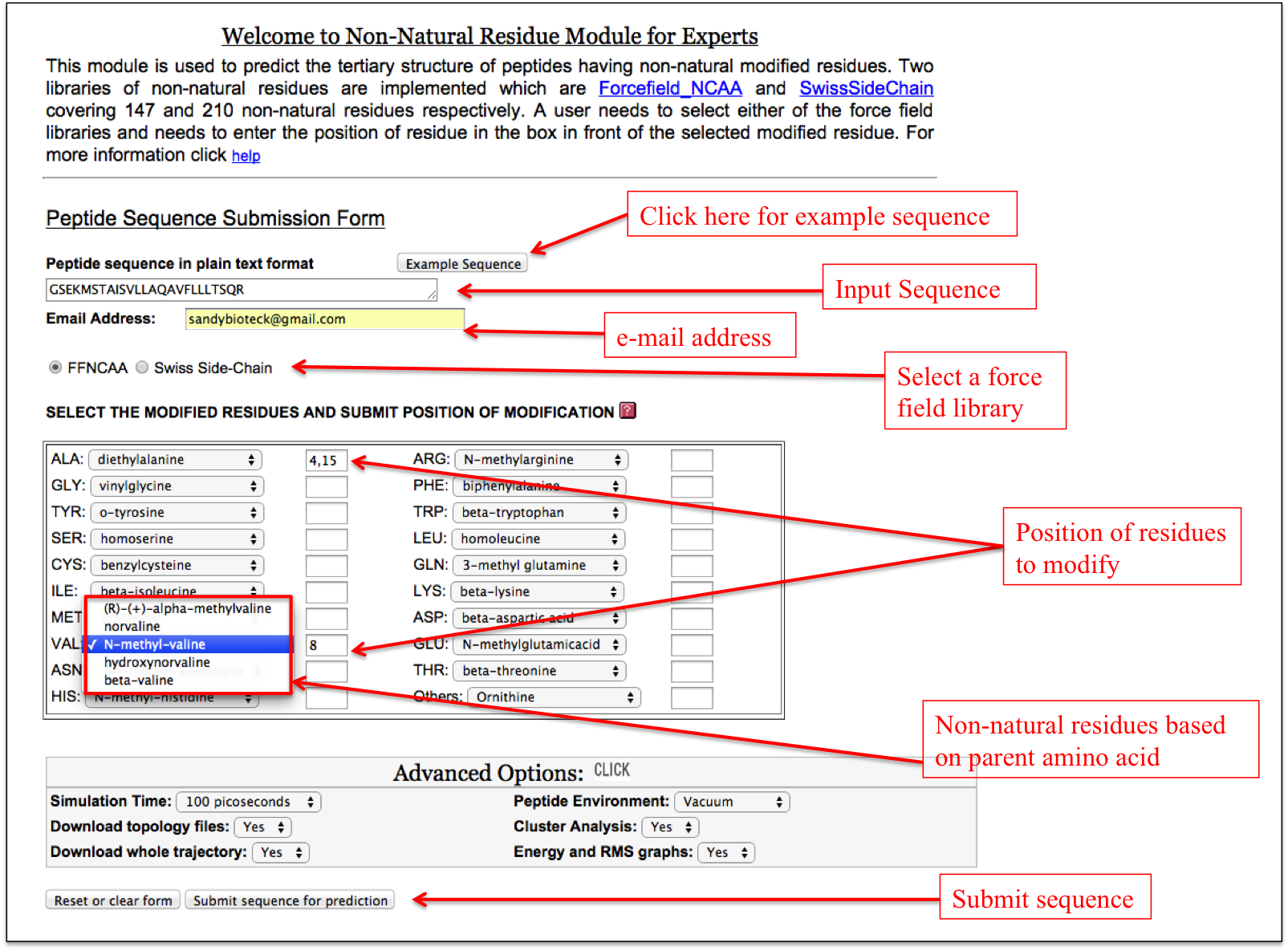
Figure 12. Labels showing how to use the “Expert” sub module of the “Non-Natural Residue” module of PEPstrMOD. |
|
Structure Modification
This facility is same as described in the Natural Peptides module of PEPstrMOD with additional option of incorporating non-natural modifications as described above in Expert sub module. Therefore, using this sub module, a user can incorporate non-natural modifications in any peptide with already available tertiary structure (Figure 13).
|
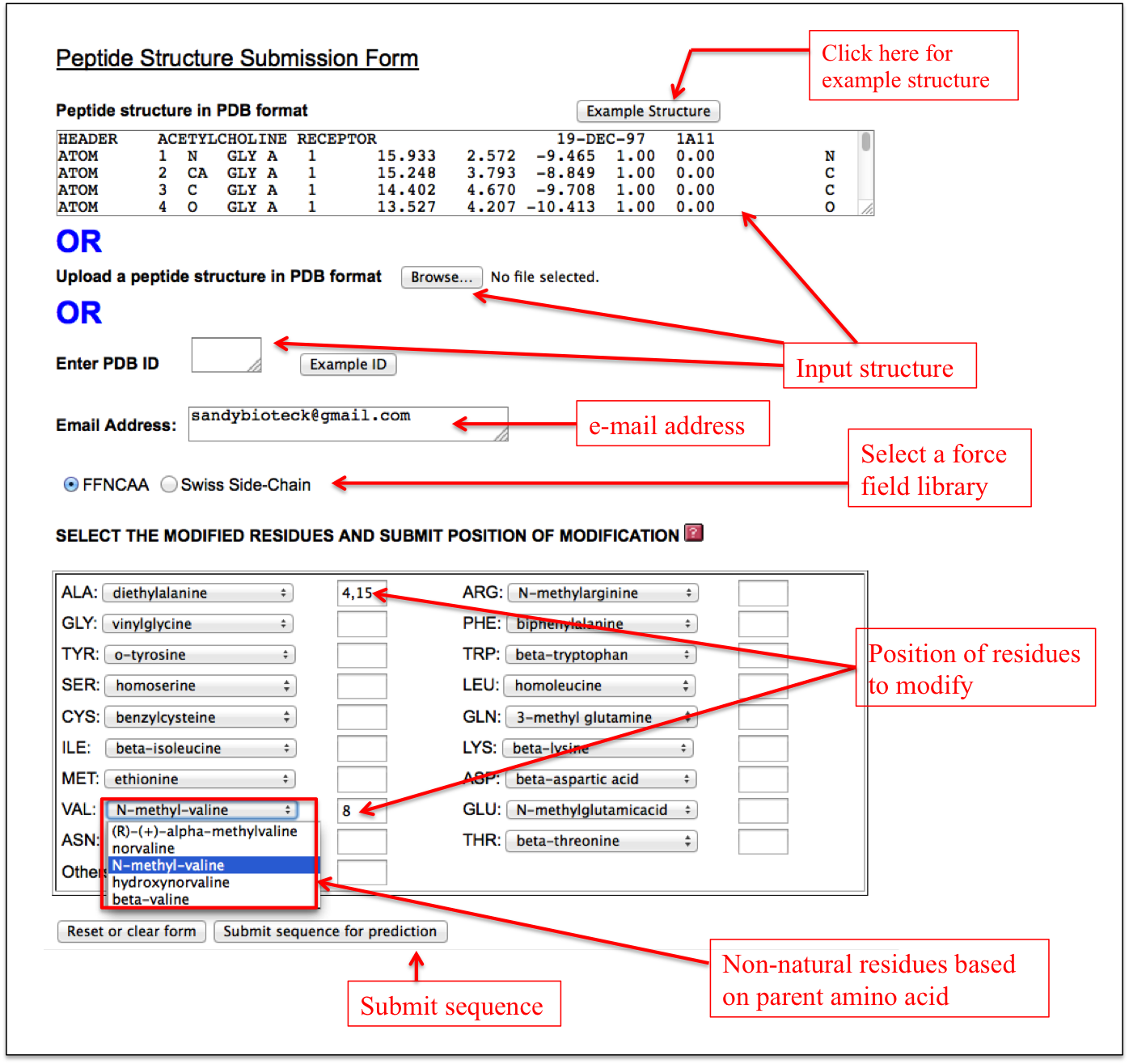
Figure 13. The snapshot of “Structure Modification” sub module of the “Non-Natural Residue” module of PEPstrMOD. The labels show how to use it effectively. |
|
PTMs of Residue |
Beginner
This sub module provides an easy alternative to post-translationally modify residues in the peptide. At first step, a user only needs to enter the peptide sequence. After submission, a user-friendly interface in the tabular form is displayed in which each amino acid of the input peptide sequence is present in one column and a drop down menu of all possible PTMs of that residue in the next column. A user can easily do post-translational modification of any residue using this facility (Figure 14A-B).
|
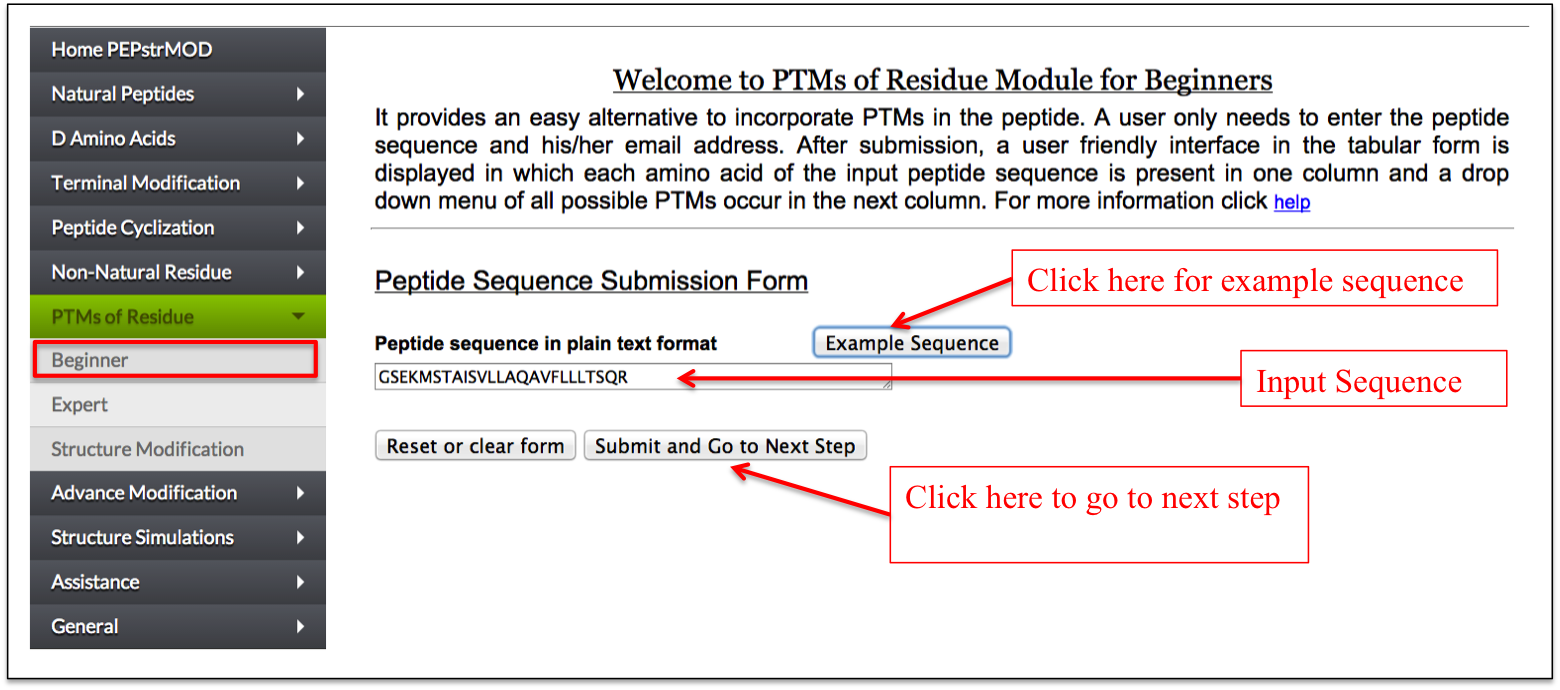
Figure 14A. Snapshot of “Beginner” sub module of the “PTM” module of PEPstrMOD with labels showing how to input the peptide sequence. The option to incorporate PTMs is provided in the next page as shown in the next figure. |
|
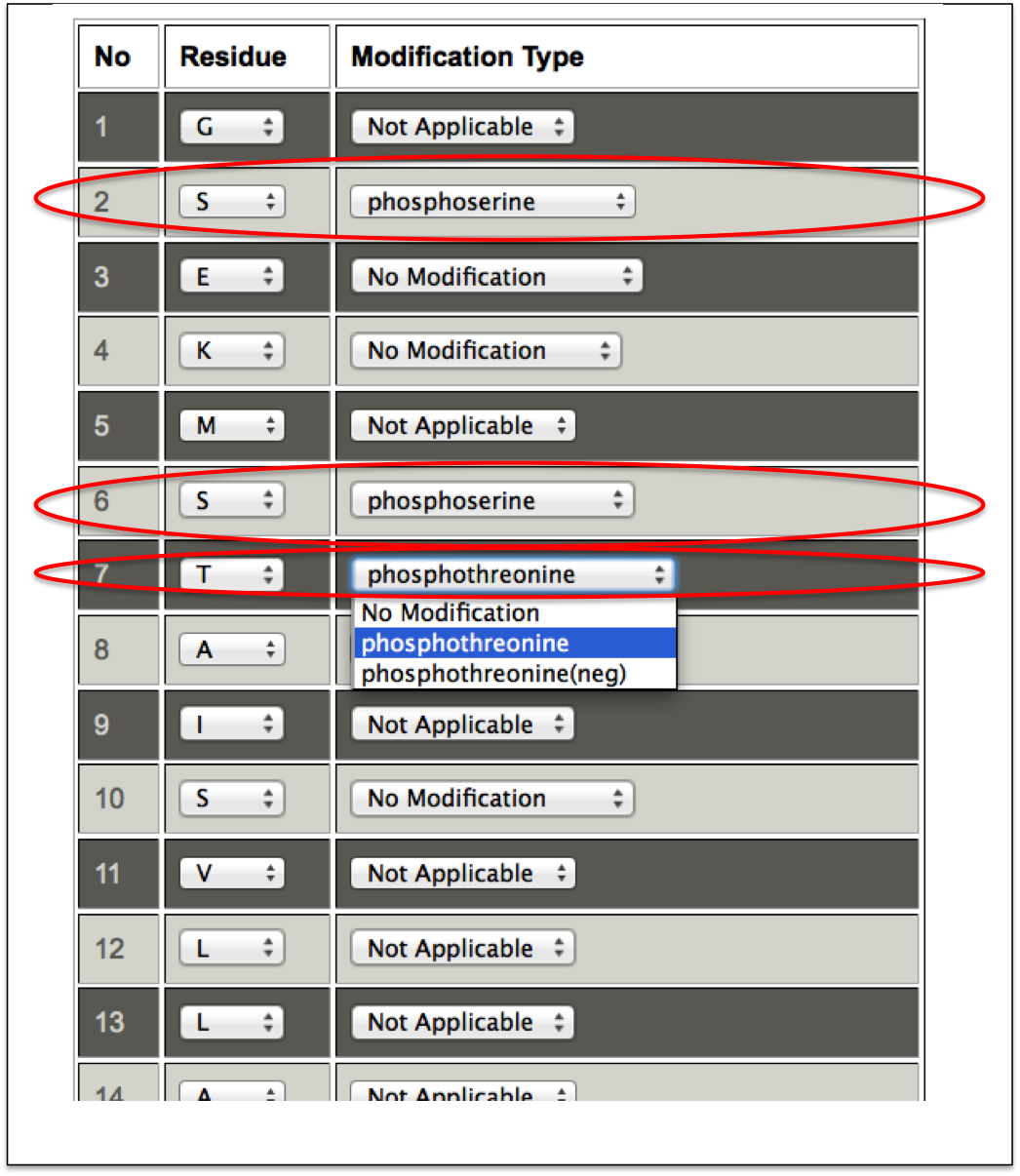
Figure 14B. This snapshot is the extension of the previous figure 14A in which peptide sequence and e-mail address was taken as input. In this step, PTMs can be incorporated by using dropdown menu in front of amino acids of the input peptide sequence. In this figure the incorporated PTMs are labeled with red ellipse. |
|
Expert
It allows a user to do post-translational modification of any residue in the peptide in a single step. For each residue a limited set of PTMs are possible. Only the possible modifications are displayed in front of the amino acids in the drop down menu followed by a text box. For incorporating a PTM, a user needs to enter the position of the amino acid in the respective text box (Figure 15).
|
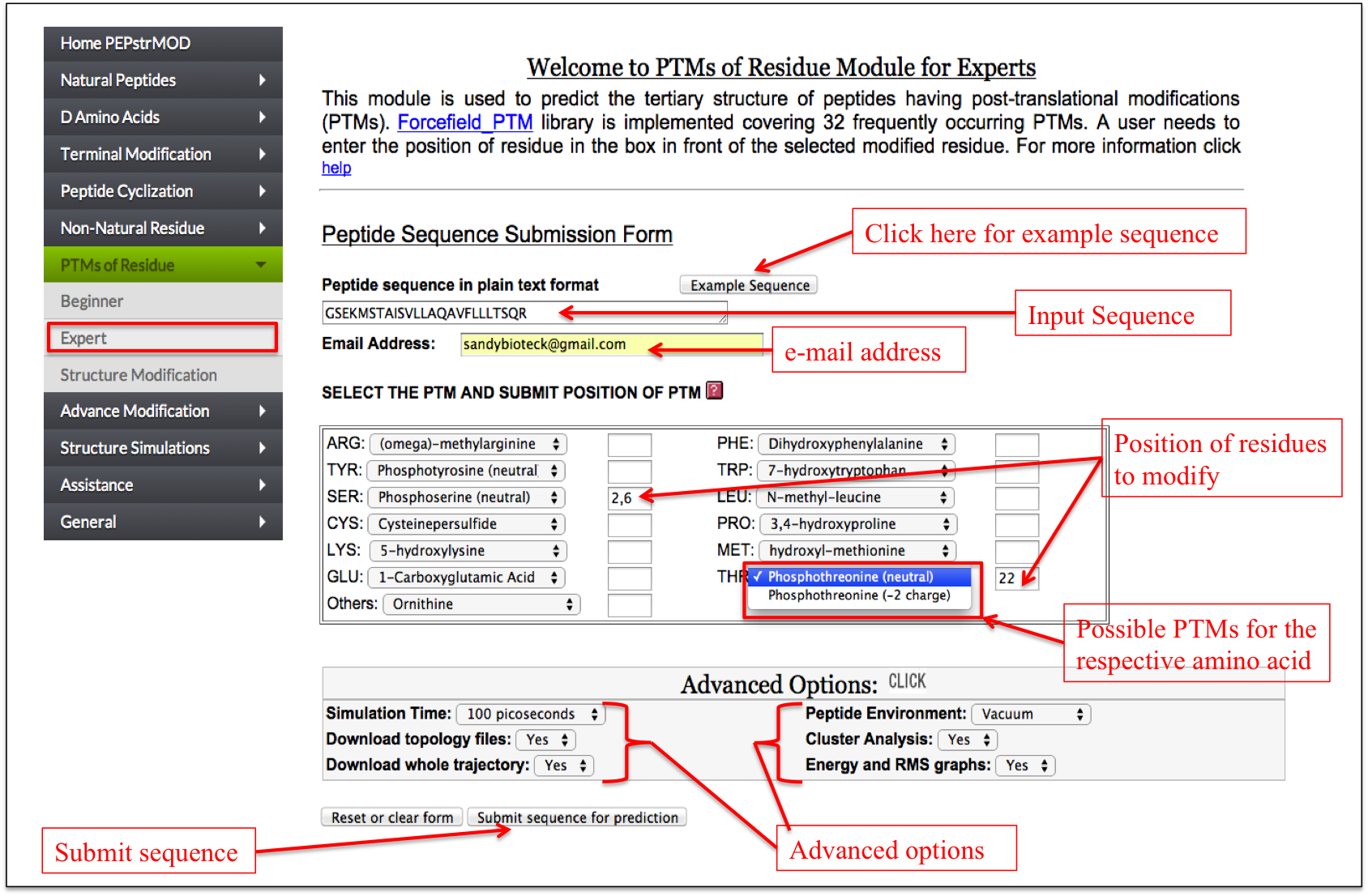
Figure 15. Snapshot of the “Expert” sub module of the “PTM” module of PEPstrMOD with labels showing how to input peptide sequence and PTMs with their positions. |
|
Structure Modification
This facility is same as described in the Natural Peptides module of PEPstrMOD with additional option of incorporating post-translational modifications as described above in Expert sub module. Therefore, using this sub module, a user can incorporate PTMs in any peptide with already available tertiary structure (Figure 16).
|
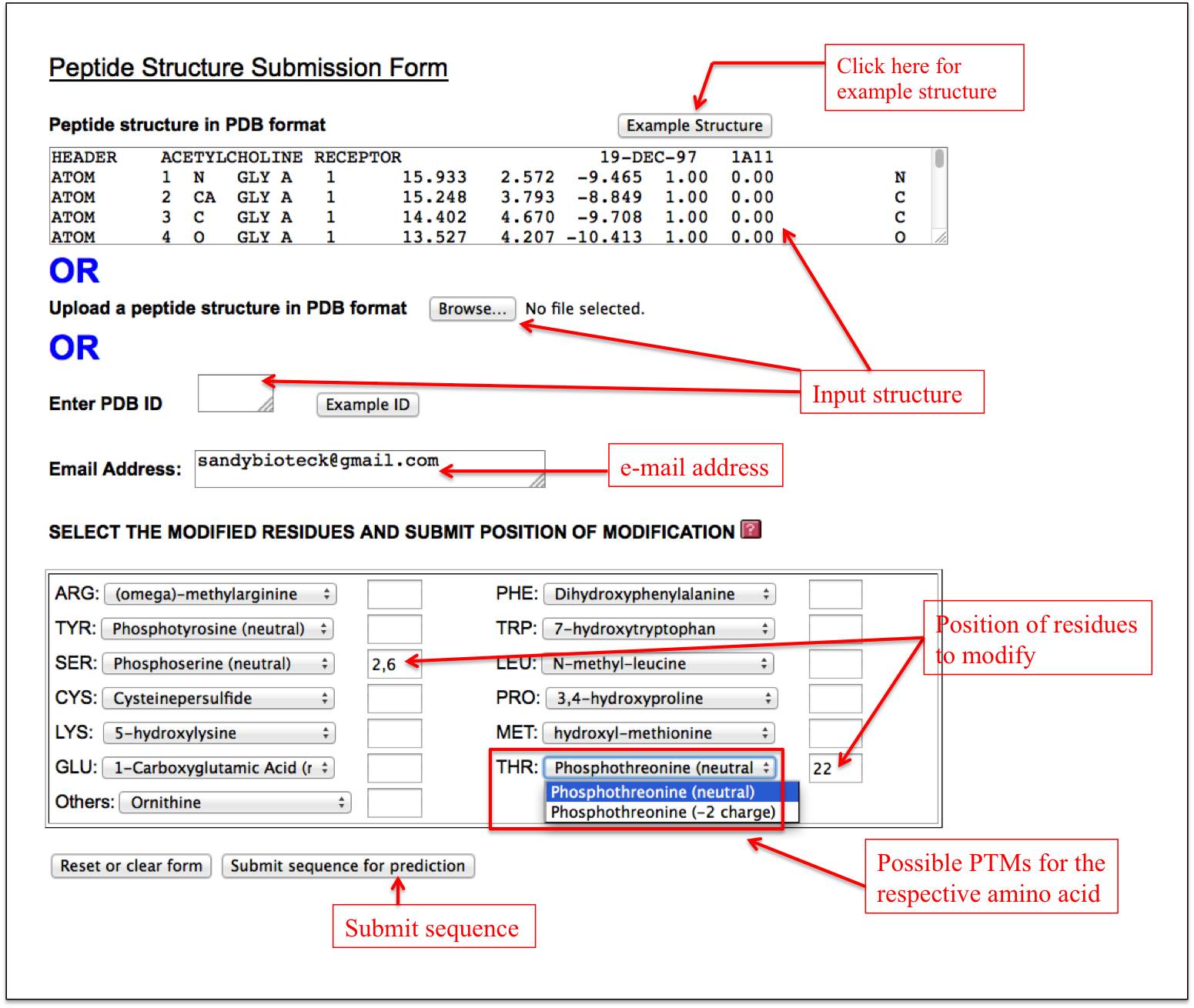
Figure 16. Snapshot of the “Structure Modification” sub module of the “PTM” module with labels showing how to input the peptide tertiary structure in PDB format and other options effectively. |
|
Advance Modification |
Beginner
This sub module provides an easy alternative to incorporate multiple modifications at different positions in a peptide. It is an amalgamation of the features of all the above modules giving an option to perform multiple modifications at different positions in a peptide. At first step, a user only needs to enter the peptide sequence. After submission, a user friendly interface in the tabular form is displayed in which each amino acid of the input peptide sequence is present in one column and different modifications corresponding to that amino acid are displayed in the drop down menu in subsequent columns. For disulfide bridge, a text box appears in front of cysteine residue. If a user wants to make disulfide bridge between 3rd and 12th cysteine, he/she needs to enter 1 in the text box in front of 3rd cysteine and enter 2 in front of 12th cysteine (Figure 17A-B).
|

Figure 17A. Snapshot of the “Beginner” sub module of the “Advance Modification” module of PEPstrMOD with labels showing how to input peptide sequence and other options. The option to incorporate different modification is provided in the next page as shown in the next figure. |
|
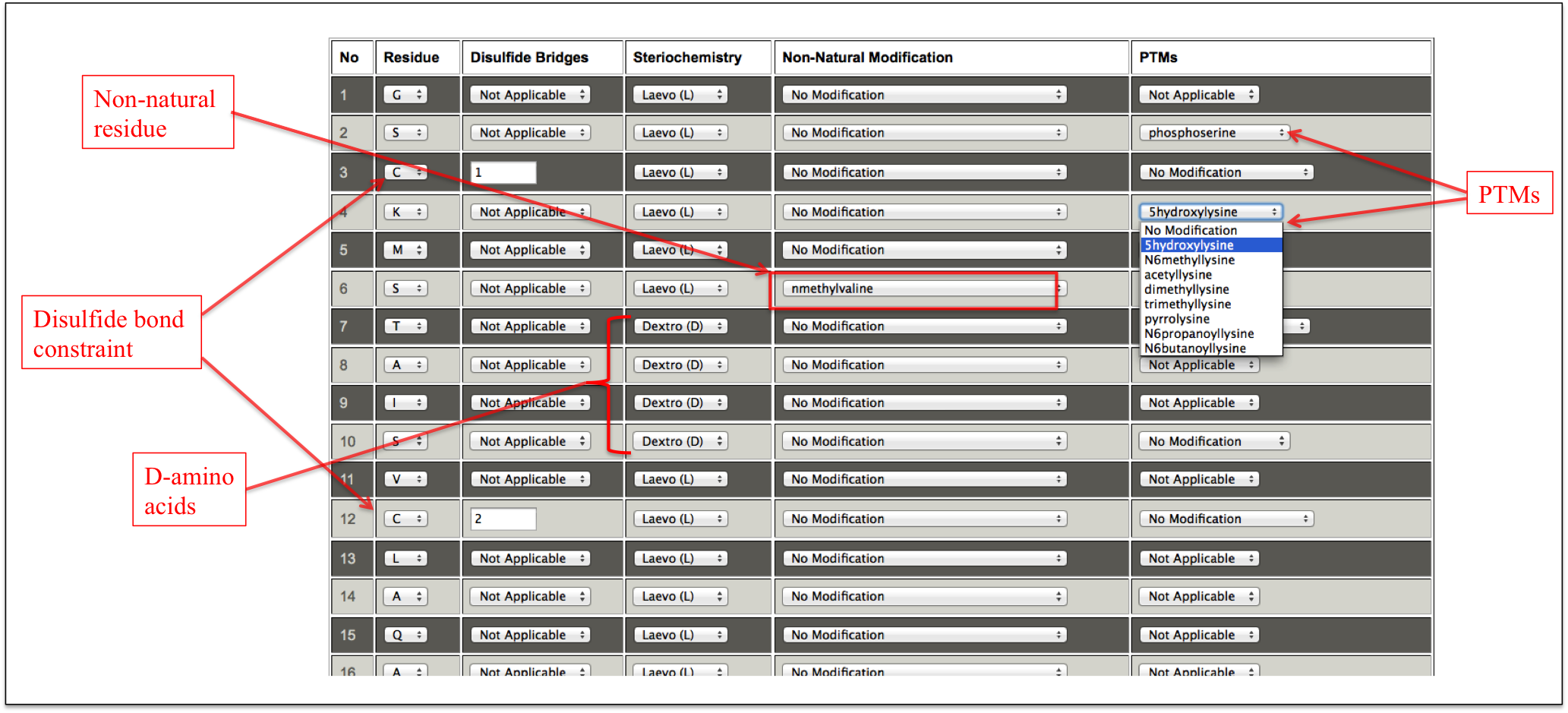
Figure 17B. This snapshot is the extension of the previous figure 17A in which peptide sequence was taken as input. In this step, different modifications can be incorporated by using dropdown menu in front of amino acids of the input peptide sequence. For incorporating disulfide bridge, a text box appears in front of cysteine residue. If a user wants to make disulfide bridge between 3rd and 12th cysteine, he/she needs to enter 1 in the text box in front of 3rd cysteine and enter 2 in front of 12th cysteine. This means that cysteine number 1 (given by user) is connected to cysteine number 2. For multiple disulfide bonds, number 3, 4 and so on can be given. |
|
Expert
A user needs to enter the sequence and his/her email address along with desired modifications. If a user wants to perform N-C cyclization, he can select yes or no. For incorporation of disulfide bridges, he/she can enter the residue positions in the box in the same manner as described in the Disulfide Bridge (S-S) sub module of Peptide Cyclization module. D-amino acids can be incorporated by making the letters small in the peptide sequence as described in D-amino acids module. Further, a user can incorporate non-natural amino acids and PTMs by entering residue positions at respective text boxes (Figure 18).
|
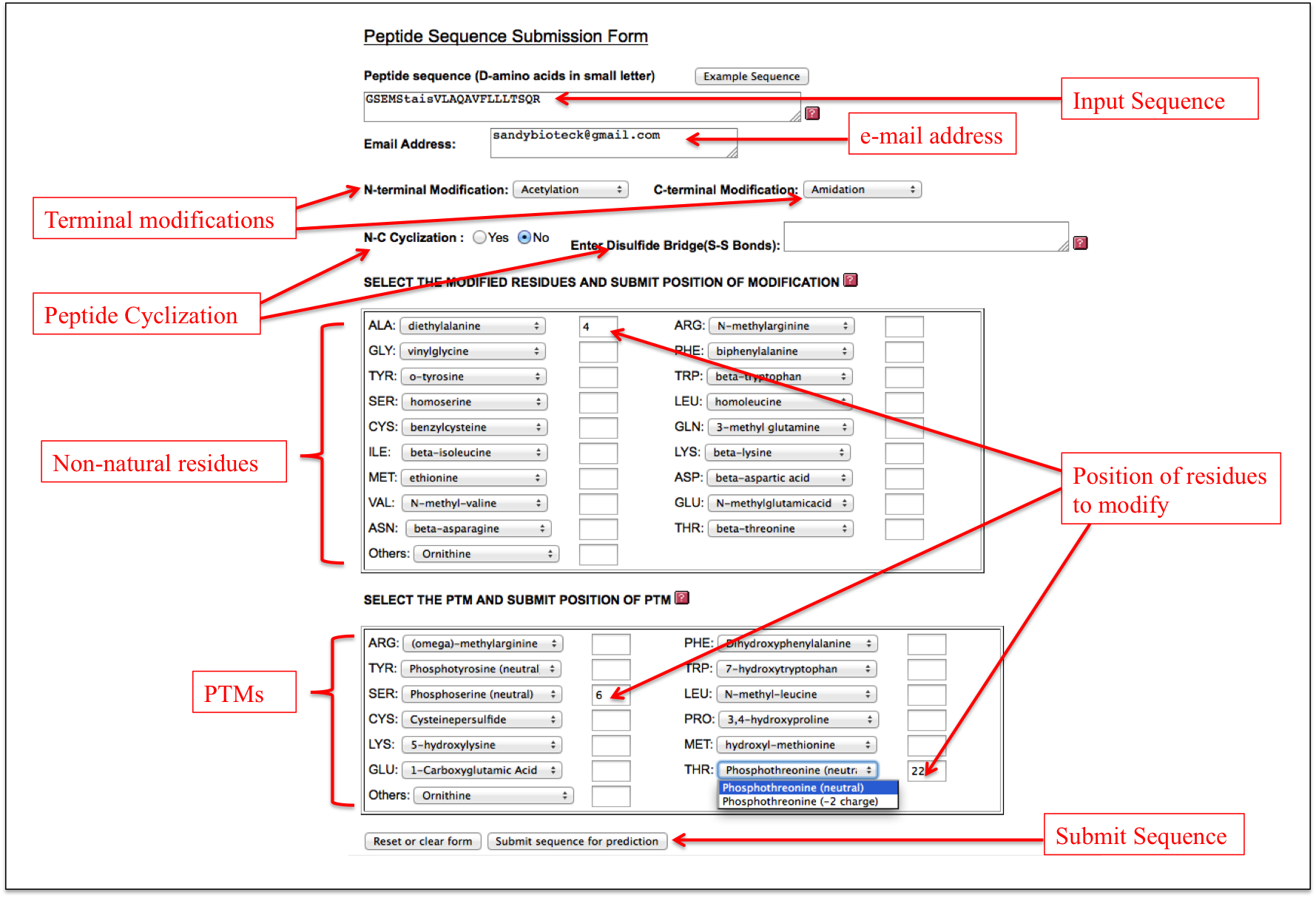
Figure 18. Graphical representation of the “Expert” sub module of the “Advance Modification” module of PEPstrMOD with labels showing how to incorporate different modifications in the peptide. |
|
Structure Modification
This sub module integrates the Structure Modification sub module of Natural Peptides module and Expert sub module of Advance Modification module. Therefore, it provides facility to the user to incorporate multiple modifications in the tertiary structure of the peptide given as an input..
|
|
Manual Modification
This sub module helps the user to open his/her structure in Marvin applet and edit the structure by incorporating any desired modification at any position and finally submit the structure. The edited structure will be energy minimized and finally given as an output to the user. This service will be useful for incorporating modifications whose force field parameters are not present in the standard force field libraries used in this study. Alternatively, a user can also edit any structure in his/her own favourate editor/software (like Pymol etc) and then open that structure here in marvin sketch and submit the structure here.
|
Structure Stimulations |
Request Token
A user needs to request for a token before using the facility of this module. A user needs to enter his/her name, email address and institute name and finally click on Submit. A token will be granted to the user, which will be mailed to his/her provided email address.
|
Simulate a PDB file
This facility allows user to perform the simulation of the peptide by inputting a PDB file of that peptide. This facility is also helpful in extending the simulations of the predicted peptide structure obtained by any of the PEPstrMOD modules. User can also give different options like Simulation Time, Peptide Environment, Cluster Analysis etc. as per his/her needs. A user first needs to request a token. The same token can be used for 3 times.
|