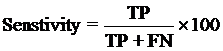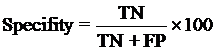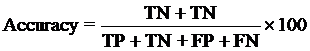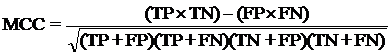Algorithm
|
In this study models were developed to discriminate molecule that have drug-like and non-drug like properties. First, we build a dataset that contain 1347 approved drugs and 3207 experimental drugs. All models were trained on above dataset and evaluated using cross-vailadtion technique. Following is brief description of techniques used for optimizing structure, calculating descriptopes, feature selection, building models etc. 1. Chemical Structure : Structure of chemicals were build and reperesnted using sdf format. Molecules in other file formats were converted into sdf format using OpenBabel software. 2. Descriptors Calculation : Descriptors of molecules were calculated using open source software PaDEL software 3. Descriptors Selection : All descriptors of chemicals are not significant, it is important to select best features or descriptors for developing prediction model. In this study we used the WEKA inbuilt algorithms (CfssubSetEval and RemoveUseless) for removing useless and highly similar descriptors. Selected descriptors were used for developing prediction models. |
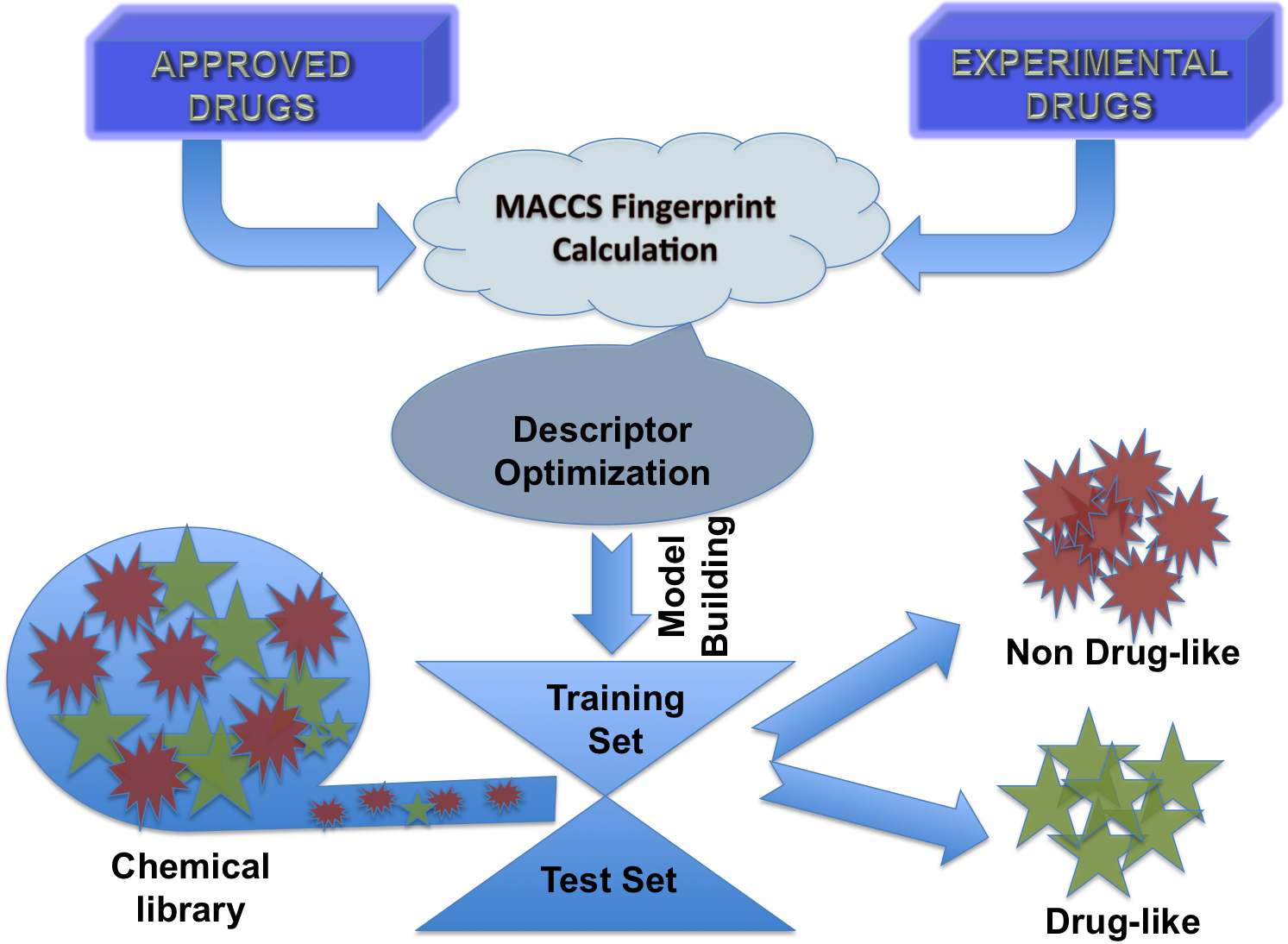
Flow Diagram of DrugMint Algorithm |
|
4. Model Development : We have used SVM-light V6.01 (Support Vector Machine) for developing classification that can discriminate drug-like and non-drug like molecules. It was observed that models based on MACCS fingerprints perform better than models based on other type of fingerprints. In this server we incorporate best SVM model developed using MACCS fingerprints. Cross-validation Techniques : The performance of all models developed in this study was evaluated using five-fold cross-validation (CV) technique. In five-fold CV, the data set is randomly divided in five sets of similar size, 4 sets used for traing and remaing set for testing. The training and testing repeated 5 times in such a way that each set is tested once. Evaluation Parameters: Following equation were used for computing evaluation parameters used in this study to assess performance of prediction models.
Search Database:
In order to provide service to community, we compute drug-like potential of moleules in ZINC and CEMBL databases. |