| Subcellular localization | Number of proteins |
| Cytoplasm | 840 |
| Mitochondria | 315 |
| Nuclear | 858 |
| Plasma Membrane | 1519 |
| Total | 3532 |
Support Vector Machines (SVM)
Evaluation of HSLpred
The performance modules constructed in this study has been evaluated using 5-fold cross-validation technique. In this technique, the relevant dataset was partitioned randomly into five equally sized sets. The training and testing was carried out five times, each time using one distinct set for testing and the remaining four sets for training. For evaluating the performance of various modules, accuracy and Matthew’s correlation coefficient (MCC) were calculated using the following equations:
![]()
![]()
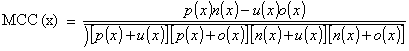
where, x can be any subcellular location (cytoplasmic, mitochondrial, nuclear, or plasma membrane), exp(x) is the number of sequences observed in location x, p(x) is the number of correctly predicted sequences of location x, n(x) is the number of correctly predicted sequences not of location x, u(x) is the number of under predicted sequences and o(x) is the number of over-predicted sequences.
Prediction
Approaches
| Subcellular localization | Accuracy (%) | MCC |
| Cytoplasm | 63.5 | 0.52 |
| Mitochondria | 46.1 | 0.52 |
| Nuclear | 76.2 | 0.67 |
| Plasma Membrane | 90.3 | 0.78 |
Dipeptide Composition was used to encapsulate the global information about each protein sequence, which gives a fixed pattern length of 400 (20 X 20). This representation encompassed the information about amino acid composition along local order of amino acid. In the case of 1-2dipeptide SVM module the best results were achieved with the RBF kernel (g=50, C=6, j=1). The SVM module was predicted with 77.8% overall accuracy which was nearly 1% better then amino acid composition based SVM module. The results obtained after 5-fold cross-validation for 1-2diepptide composition are shown below:
| Subcellular localization | Accuracy (%) | MCC |
| Cytoplasm | 58.3 | 0.52 |
| Mitochondria | 48.3 | 0.52 |
| Nuclear | 80.2 | 0.71 |
| Plasma Membrane | 93.4 | 0.80 |
| Subcellular localization | Accuracy (%) |
| Cytoplasm | 56.9 |
| Mitochondria | 40.6 |
| Nuclear | 68.2 |
| Plasma Membrane | 92.0 |
HYBRID SVM module
This module uses the complete information about the protein that is amino acid composition, dipeptide composition and evolutionary information of PS-BLAST output. SVM was provided with an output vector 425 dimensions that consisted of 20 for amino acid composition, 400 for dipeptide composition, five for PSI-BLAST output. The performance of this module was better then any other individual feature based module. This hybrid module with the RBF kernel (g=50, C=2, j=1) was able to achieve overall 84.9% accuracy. The results obtained for four types of subcellular localization are shown below:
| Subcellular localization | Accuracy (%) | MCC |
| Cytoplasm | 75.4 | 0.67 |
| Mitochondria | 69.8 | 0.68 |
| Nuclear | 82.4 | 0.79 |
| Plasma Membrane | 94.8 | 0.89 |
Other SVM modules
We have also constructed other various types of dipeptide SVM module (1-3, 1-4 and 1-5) and hybrid modules . The accuracy obtained for the SVM modules are shown in the graph below
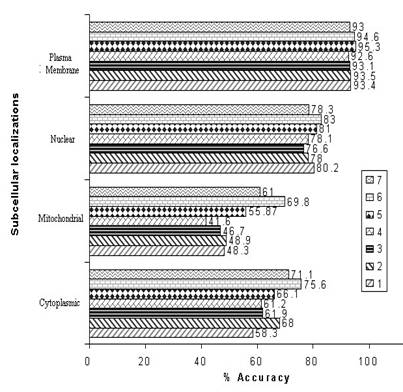
where
1 is 1-2dipeptide composition (D)
2 is 1-3dipeptide composition (E)
3 is 1-4dipeptide composition (F)
4 is 1-5dipeptide composition (G)
5 is hybrid2 SVM module (H=D+E+F+G)
6 is hybrid3 SVM module (Amino acid +H+ PSI-BLAST)
7 is a cascade SVM
RELIABILITY INDEX
The reliability index (RI) is a commonly used measure of prediction that provides confidence about a prediction to the users. The RI assignment is a useful indication of the level of certainty in the prediction for a particular sequence. The RI was assigned according to the difference between the highest and second highest SVM output scores. We have also computed the reliability score of our prediction method based on the hybrid approach using the following equation
![]()